
|
The following excerpt comes from:
P.Rotkiewicz, W.Sicinska, A.Kolinski, H.F.DeLuca, "Model of three-dimensional structure
of vitamin D receptor and its binding mechanism with 1-alpha,25-dihydroxyvitamin D3",
Proteins, 44, 188-199 (2001)
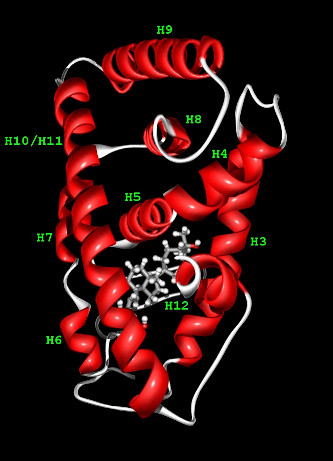
The Vitamin D Receptor is a 423-residue long protein consisiting of two domains: DNA binding domain
and ligand binding domain (LBD), the latter one pictured to the right.
The VDR belongs to the nuclear receptor (NR) superfamily including receptors for the steroid, retinoid and thyroid hormones.
While DNA binding domains (DBD) of these receptors are generally highly conserved (sequence identity over 40%), ligand binding domains
(LBD) exhibit limited similarity. Crystal structures of the nuclear receptors reveal well conserved architecture (11 or 12 antiparallel
helices composed in three layers) of the LBD.
To gain a better insight into the binding mechanism of the ligand to the receptor one needs as accurate a structure
of the VDR as possible. Fortunately, high-resolution structures of five highly homologous proteins are already known. All of them
have been used as a multiple template for comparative modeling of the VDR structure. Since the template proteins are structurally
very similar, it is expected that the resulting molecular model of VDR should be very close to the true (unknown) structure.
Having putative structure of the VDR receptor and knowing the experimental structures of receptor-ligand complexes of five homologous
proteins allows to predict with relatively high fidelity the set of residues that constitute the binding pocket of the VDR.
It was achieved by a conservative analysis of the multiple sequence alignment of the five template proteins and the VDR sequence.
Using the VDR structure obtained from comparative modeling we performed automatic docking of the six distinct conformers
of the hormone into the putative binding pocket. Docking procedures enabled conformational flexibility of the ligand, however the receptor
structure was assumed fixed. The obtained structures of the receptor-ligand complexes were then critically evaluated using the criteria
of plausibility, internal consistency and the ability to explain experimental facts.
Computational part of this work consisted of several steps. First, crucial for the entire procedure, is the identification
of the template for the comparative modeling and generating of appropriate multiple sequence alignment.
Five highly homologous proteins of known structure were identified. These were subsequently employed in VDR model building
using an automated molecular modeling program MODELLER. The resulting molecular model was refined using molecular dynamics
and SYBYL force field. Finally, six forms of 1 ,25-(OH)2D3 hormone were inserted into the VDR binding pocket by means
of flexible docking procedure. For each form of the ligand the docking procedures were performed several times, starting
from various arbitrary chosen initial positions (and conformations) of the ligand in a vicinity of the binding pocket.
|

|
|
|
|
|
